
Open Letter to Scott Alexander
It's ok to change your mind
Dear Scott-
I was a bit surprised to see your old arguments that Covid came from the wildlife market resurface in the Comments on Richard Hanania’s substack criticizing a sleazy fascist theorist. (Don’t ask how I stumbled onto it.) You have some intensely devoted followers who think that you are the most reliable source on this issue. You may not particularly desire that sort of cult following, but it’s there. I think that the impact of your important points on Covid public health policy is dragged down by the weak arguments about the origins.
So I can’t resist showing both line-by-line and then with regard to general methods how you’ve screwed up the Bayesian argument. As a psychiatrist, you may be well qualified to judge how insane my fantasy is that you might change your opinion when shown to be wrong. Short of that, it might help to let some of your followers know that none of us should be trusted too much– there’s no reliable substitute for really following the arguments.
Pekar 2022
I’ll start with your comments on the Pekar et al. 2022 paper (P2022) on the “The molecular epidemiology of multiple zoonotic origins of SARS-CoV-2“, whose central claim was that there were two separate successful spillovers from wildlife to humans. That two-spillover claim turns out not to be extremely relevant to the origins question, but the paper was so thoroughly wrong that it provides a good limiting case to see how reliable someone’s analysis is and to see if they have a one-sided slant. So let’s look line-by-line at your analysis of whether it was “debunked”.
Scott: “Pekar’s paper on the two lineages originally estimated 99-1 odds of double spillover.”
Incorrect. The original published paper had 60-1 odds.Scott: “Someone found a coding error that reduced it to 6-1 odds, Pekar admitted the error, and the paper has been updated.”
Incorrect. Someone (Angus McCowan, then writing pseudonymously on the pubpeer post-publication review site and now on arXiv) found three significant coding errors that later were acknowledged by the P2022 authors. These reduced their published corrected odds to 4.3-1, not 6-1. Since they had originally defined 10-1 as the cutoff for “significance” they just quietly changed that cutoff to 3.2-1 in the corrected version to keep the results “significant”. Meanwhile McCowan found a few other minor coding errors and reran the code enough to reduce statistical imprecision, getting 3.5-1.Scott: “Other people have made other criticisms which I haven’t investigated in depth and am agnostic on.”
Strange. The clearest big remaining error, also put on the same pubpeer thread by the same McCowan, was not some detail. It was a deep, extreme error in basic Bayesian logic. They compared a likelihood for the two-spill hypothesis to produce some broad range of outcomes with the likelihood for the one-spill hypothesis to produce a radically narrower range! Specifically the one-spill was required to produce two polytomies differing by at least two nucleotides with the smaller one having at least 30% of the overall phylogeny while putting no constraint at all on the nucleotide difference or relative size for their favored two-spill hypothesis. McCowan describes this on arXiv and I translate to easier terms and summarize some of the other big issues here.
Just to make sure that crucial point is clear to anyone unfamiliar with the basic methods here’s an example. If one were to update the odds for deciding which of two suspects committed a burglary, it would not be correct to use the ratio P(drives car|suspect 2)/P(drives blue Toyota|suspect 1). That gives a completely bogus factor pointing toward suspect 2. That would be a fundamental error in logic, precisely analogous to the Pekar et al. error. That type of extreme error in basic logic is grounds for retraction. How can anyone claiming to use Bayesian techniques be “agnostic” on that?
Scott: “The argument for B earlier than A is that it infected twice as many people and has more genetic diversity. It’s possible these things happened by chance and A preceded (and mutated to) B.”
Red herring. The number of people infected has almost no significance because in the early days of a pandemic the statistical fluctuations in transmission are huge, particularly given spotty environments. The interesting molecular clock peculiarity, i.e. genetic diversity, was noticed much earlier but was not even used in the P2022 Bayes factor for comparing the hypotheses. Usually the more ancestral lineage would tend to show more diverse descendants, but the reverse happened here, which is a bit unlikely. For the two-spill hypothesis the same sort of chance is required to get the molecular clock out-of-synch but just before the spills. The less ancestral lineage would have to have spilled first, then by some improbable event the more ancestral lineage spilled later without having any further mutations in the meantime. I haven’t seen any even toy model calculations estimating which of these moderately unlikely versions is more unlikely.
Scott: “In that case, I still think the most likely scenario is that A was released at the wet market, infected a customer or two, mutated to B, and infected a vendor. A then spread among the neighborhoods near the market, and B spread among market vendors.”
Circular. That’s just restating your conclusion.
Scott: “Several people accused Pekar of ignoring intermediate lineages. Peter addressed this by finding these were mostly sequencing errors. There’s a very new paper about potential intermediate lineages which might change this debate; my provisional assessment is that it’s boring but I’m waiting to see if other people have more thoughts on it.”
OK. Low detection and sequencing rates early on mean that it was hard to find early intermediates. Simply using the Pekar data alone but fixing the basic logic error makes the odds favor a single spillover even without the intermediates found by Lv et al..
So that’s probably enough specifically about P2022 and the two-spill hypothesis. Reversing the odds doesn’t say a great deal about the origin of covid because P2022 was never all that relevant, despite the fancy title and press-release hype. What it should do, Scott, is to call for some introspection about whether you are really calling this in an objective way. Almost every sentence you wrote about it was wrong or misleading in a consistent direction.This later passage may give some feel for why that happened, although not at a depth-psychological level.
Scott: “In general, I find claims about “debunking” annoying even when they’re made by Important People who theoretically have the authority to make pronouncements. I think they’re even more annoying when they’re made by self-styled rebels who admittedly disagree with the scientific consensus.”
”Debunk” is an irritating and overused word, agreed, but in this case the problem with “debunked” is that it would be an understatement. The rigorous demonstration of a deep logic error in P2022 was done by Angus McCowan, a patent expert who claimed no authority but mathematical logic. With the French National Academy of Medicine, the German DNI, the US FBI, CIA, DOE Z-division, National Center for Medical Intelligence, the Brookings Institute, RAND, and the UK MI6 all at least somewhat leaning toward a lab leak, at what point does the definition of annoying ”self-styled rebels who admittedly disagree with the scientific consensus” flip directions? That’s not a robust way of framing the issue and deciding what’s reliable.
Worobey and Ascertainment Bias
Your net result, a factor of 17 favoring a market origin, depends entirely on a Bayes factor of 5000 from “First known cases in wet market” and “First A near market. (When some other market-specific factors are included, you end up with a market-related factor of 2200.) This factor rests entirely on the Worobey et al. paper (W2022) and Peter Miller’s retelling of its central claims, as recounted in your blog.
W2022 shows that starting in mid Dec. 2019 a majority of the ascertained cases hit people who’d been to HSM. So HSM was definitely a superspreading location. The question is whether the substantial fraction of cases that were not linked to HSM somehow also traced back to there but by some missing steps. The alternative is that some cases were kicking around for a while before the HSM cluster took off. W2022 shows a remarkable map in which the home addresses of most of these unlinked cases (represented by the yellow dots and blue areas below) cluster around almost exactly the HSM. They point out that the unlinked cases cluster significantly closer to the market than do the linked cases. The map shown here, assembled by Daniel Walker, combines the published W2022 map of the unlinked cases with a separate W2022 map of the linked-cases, which was posted only in a separate supplement. The linked cases not only are more spread out but also, unlike the unlinked cases, cluster away from the market.
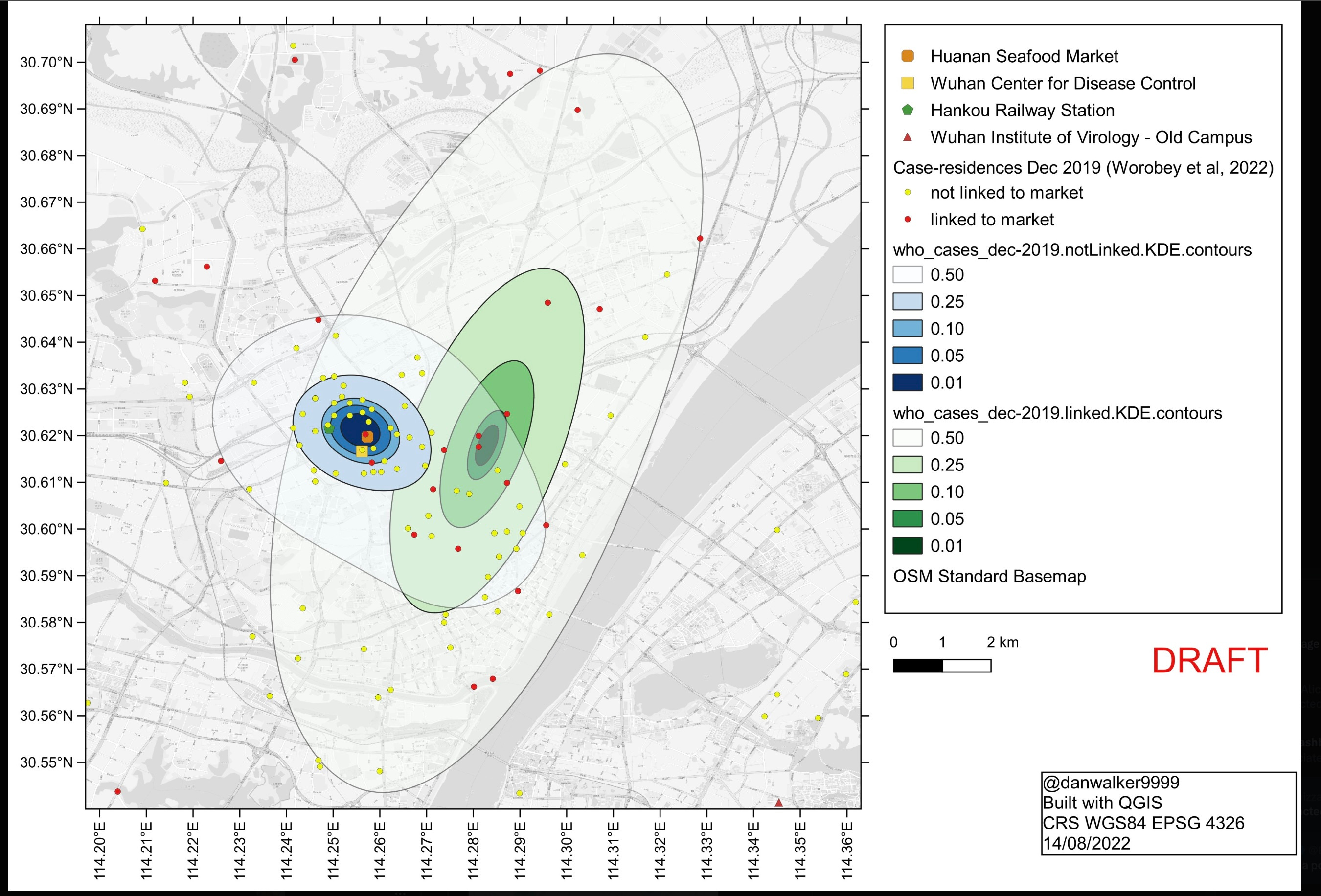
Many people have pointed out that the history of how unlinked cases were found would preferentially have ascertained ones in people who lived near HSM. The question is whether that’s a big effect. Here I won’t rehash everything that’s already been said about this but will just focus on my contribution and your response.
Scott: “What is the Weissman paper ….? It argues: if the pandemic started at the market, each seemingly non-market-linked case must ultimately derive from a market-linked case. Therefore, we should expect non-market-linked cases to require more steps than market-linked cases. Therefore, they should be further away. But if we look at the map above, we see that not-market-linked cases are closer to the market than market-linked cases. So something must be wrong, and that something might be ascertainment bias.” (at least this is my interpretation of Weissman’s argument, which is more mathematical; read the paper to make sure I’m getting it right).”
Yes, that’s how it goes. It’s not really much more mathematical than that, although I use the standard statistical term “collider stratification bias” to help readers find literature on the effect. It didn’t require me looking at the map. I simply quoted a statistic that W2022 made a point of featuring without noting that it had the wrong sign for the model they used and the right sign for the competing ascertainment bias model.
Scott: “This is a weirdly spherical-cow view of an epidemic, worthy of a physicist. It’s easy to think of reasons the linked-cases-should-be-closer rule might not hold. For example, suppose that on their lunch break, market vendors go have lunch at restaurants surrounding the market. They infect people in these restaurants, who then infect their friends and family. But these people never went to the market themselves. Now there are a bunch of non-market-linked cases immediately surrounding the wet market.”
You’re right about the “spherical cow” part, and that real cows aren’t spherical. You can always make up stories about why the HSM neighborhood was extra-special and an excellent place for there to be a cluster. It could well be. For example, after two months in Beijing with no ascertained local transmission, there was a sudden outbreak. Out of all possible places in that huge city, it happened at the Xinfadi wet market and environs. That does not make Xinfadi the spillover site. Similar abrupt market outbreaks happened in other large cities, so Xinfadi was not a rare fluke. Markets seem to be great locations for initial big outbreaks.
The problem was that the original W2022 was a pure spherical-cow paper itself, arguing that the HSM cluster must indicate the spillover site because ultra-simplified models of other sources didn’t fit the cluster. Miller’s model of uniform spread at a fixed exponential rate in an unstructured population, from which you obtain your huge Bayes factor, was even more obviously a spherical cow. What S. Zhao et al. wrote about P2022. applies also to the other toy models: “… they assumed that viruses spread and evolve without population structure, which is inconsistent with viral epidemic processes with extensive clustered infections, founder effects, and sampling bias.”
Worobey teamed up with Débarre (DW2024) to try to answer my paper. They argued first that W2022 didn’t really mean it about the unlinked cases clustering much closer to HSM since W2022 used an oversimplified model lacking overdispersion which leads to more clustering. So it could have just been a statistical fluke after all, which they tried to support with an explicit model. One problem with that is that none of W2022’s calculations that were supposed to show that the clustering implied that HSM must have been the source used that overdispersion. With DW2024’s explicit model, we (Jon Weissman helped) could finally put some quantification on how badly the data fit the model. It turned out that unless the distances that unlinked cases traveled from infection to home were much smaller than the corresponding distances for linked cases and the distances from infection site to infection site in the unlinked steps were really small compared to the distance to home, it was highly improbable that unbiased sampling would give the reported results. DW2024 also argued that special features of the HSM neighborhood could have given that clustering. Could be, though we showed that the features have to be pretty extreme. What they fail to note is that in the presence of such strong population structure the whole W2022 paper falls apart. Andrew Levin’s model makes a first-order pass at including the population structure in a standard way by including density and reverses the Worobey conclusions.
Scott: “But also - of all markets in Wuhan, Huanan sold the most weird wildlife. Suppose someone in the boonies gets a craving for raccoon-dog one day, their local convenience store doesn’t have it, so they hop on a bus and go downtown to the city’s main wet market. Then they get infected with COVID. Now there’s a wet-market-linked case in the boonies.
In other words, we should expect two modes of spread: general geographic diffusion from the epicenter, and people from far away who made specific trips.”
Nice story. There’s real data from New York showing that local clusters popped up after less-local spread along transit lines. As long as we’re making up stories, maybe one of the gang at WIV with the sniffles took Metro Line 2 over to HSM to pick up some shrimp. Then the rest plays out pretty much along the lines you gave. Or maybe a collaborator at the CDC viral lab was pre-symptomatic and walked over to pick up snacks for an opening day party on the day the CDC opened its new lab 280 meters from HSM. That was about a week before the first reported HSM cases. Plays out pretty much the same way.
Scott: “So I don’t think Weissman’s paper proves anything, and I think the general pattern of blue and orange dots suggests ascertainment bias wasn’t playing a role.”
Your subjective guess is based on what? How the hell would you tell if the clustering comes from ascertainment bias? What’s the actual argument?
Meanwhile Levin has tried analyzing that pattern using standard spatiotemporal methods ignoring ascertainment bias. He obtains extremely large Bayes factors weighing against the simple Worobey model and large Bayes factors supporting a source on the south side of the Yangtze rather than just the north-side HSM. I don’t believe his models are especially reliable. The point is (see below on hierarchical Bayes) that an extreme Bayes factor can’t be correctly derived directly from some model that’s maybe sort of right used on data that is maybe not too biased. The correct Bayes factors are limited by the “maybe’s” not by the extreme factors that the toy models give.
Scott: “So why does George Gao say that there was ascertainment bias? I looked for the direct source of the Gao quote and couldn’t find it; if someone else is able to, please let me know, since I’d be interested in exactly what he thinks about this.”
Here, in an interview with the BBC (start at 23:40) George Gao, head of China’s CDC, acknowledged that there was intense ascertainment bias so that “maybe the virus came from other site”. [The audio is unclear whether the word is “site” or “side”, but Gao later told B. Pierce that he meant ”site”, less directly suggestive of WIV than “side” would be.] (That’s cut and pasted from my long Bayes blog, in which you would only have needed to search for “Gao”.)
Hierarchical Bayes: “Maybe” means “Maybe”
Your Bayesian analysis ends up favoring Z over L by a factor of 17. Even at first glance what stands out in that analysis is the huge factor of 2200 from the HSM-specific data, mostly simply from the first official cluster being at HSM, with smaller adjustments for other data. Drop that modeling-based net HSM factor of 2200 and your odds go to 130/1 favoring L. Does anything like that factor of 2200 belong in a proper calculation?
Let’s look at some familiar problems. Say that most of the evidence in a criminal trial points to suspect A but there’s a DNA sample from the scene that points to B, who unlike A happens not to be a friend of the police. The standard odds factor for that DNA match is enormous, say 1,000,000,000. Should you conclude with near certainty that B is guilty? That DNA is really strong evidence, if.
If the cops didn’t fake it. They don’t usually but sometimes they do.
And if the lab actually did the test, which they usually do. Sometimes they get lazy or lose a sample and just report the desired results.
and if nobody screwed up handling samples in the lab and contaminated the crime scene sample. etc.
That DNA is evidence but not billion to one evidence.
How much evidence? For that you need hierarchical Bayes. There’s a tree of branching possibilities. Was the lab sent the real sample or not? Did they really test it or not? Did they accidentally mix up samples or not? The easiest and in some sense least important step is the last one- how likely would it be for that DNA to come from somebody other than B? The odds come from the probabilities of going down the right branches at every fork to even get to that last step.
Here’s another famous example. Remember when the Hubble telescope first went up it was out of focus, and required a subsequent visit to do some space optometry? How could that have happened with such a big project? The focus had been tested with two devices, one rough one and one very precise one. They didn’t agree so the precise one was used. The odds of it being off by much were way lower than the odds of the crude device being off. Of course, those extreme odds did assume there wasn’t a highly reflective little spot in the wrong place, where the anodization had been chipped. Whoops.
Here’s a particularly relevant example of a mistake I made. I initially estimated P(CGGCGG|Z) to be ~1/1500, under the plausible assumption that coding within the FCS region would resemble other coding in the virus if it had evolved naturally. (It later turned out that within that homogeneous model, the results should have been even more extreme since less than 1/10,000 RR pairs are coded CGGCGG in Asian betacoronaviruses.) I had skipped the first step in the hierarchical analysis by assuming that nothing distinguished the RR in the FCS much from all the other natural RRs. In fact there are other lines of evidence that the FCS arose as a single-step large insert for which the probability of RR being coded CGGCGG in nature is more like 1/50. Even though such recent large inserts aren’t a big fraction of viral sequences (if they were the CGGCGG coding overall couldn’t be so rare) they provide a way around the extreme odds that I’d initially used for that factor.
The general idea is that for any observation the conditional probabilities of that observation under some hypothesis involve judgements about the accuracy of the observation, the sufficiency of the form of the model used to calculate outcomes for the hypothesis, and the plausible unknown ranges of parameters that go into the model. These are not optional little tweaks but lie at the core of the method. A gambler who ignores them will go broke.
Other than Saar Wilf’s “rootclaim” method the participants in the debate you watched seem to have just brushed past this required step, at least in the few hours (focused on parts that Miller had said were important) that I could put up with watching. (Subsequent write-ups and discussions make it clear that this step was indeed ignored in practice.) For small Bayes factors this step can be skipped but for big ones obtained from models of dubious data these caveats are no longer minor tweaks but become the main factors determining the conditional probability of the observation. The precise outcome of the extreme model becomes the minor tweak. Rootclaim uses a sort of reduced-instruction-set version of hierarchical Bayes methods. They don’t integrate over distributions of uncertainties in various parameters but they do truncate probabilities obtained from a model at some estimated probability of the model being qualitatively right. They might be more convincing if they just said they were doing a simplified version of standard hierarchical Bayes rather than hyping their method as something special. By far the biggest difference between their results and the others (excluding Peter’s trolling) simply comes from their truncating the extreme HSM location factor on the grounds that it isn’t especially reliable.
So let’s turn to your big factor from the HSM cluster. It can be pretty significant if the early case data were fully reported. For that to be true we need all of:
(0) Unintentional collider stratification bias isn’t important.
(1) The reasons later given for dropping the initial unlinked cases from the early Chinese reports need to be valid.
(2) No other improper data-editing was done.
(3) The reliable-looking reports that SC2 showed up in wastewater samples from Milan and Turin on Dec. 18, 2019 are wrong.
(4) The many different reports from Western sources of hearing about a major new respiratory disease in Wuhan by mid-December (see e.g. 1:50 here) were all false for one reason or another.
(5) Gao, Pekar 2021, and many other prominent scientists were wrong in their claims that there was probably spread before the HSM cases started even though these are scientists who deny the likelihood of a lab leak.
Each one of those conditions might turn out to be true, but the correct odds that you get from the reported HSM cluster have to be less than the odds that every one of those conditions is true. Does anyone in their right mind think that the odds that all of them are true are greater than 2200-1? Even at 100-1 your odds would start to favor L.
What would realistic odds from the existence of the HSM cluster be? Empirically, looking at the occasional big market outbreaks in other big Asian cities, maybe 20-1. (Yuri Deigin tried to make that point in the debate with Miller but maybe over-emphasized mechanistic reasons for non-spillover market outbreaks rather than the simple empirical result that they aren’t rare.) Once those realistic empirical odds are combined with other HSM-related odds (especially Wuhan’s low fraction of Chinese wildlife trade compared to its population fraction, negative swab nucleic acid correlations with hypothetical hosts unlike real animal coronaviruses, lack of cases among wildlife vendors) the HSM-specific odds fall below the background odds for generic zoonosis by unspecified routes.
My old error
After the 2001 attacks on the World Trade Center and the Pentagon the U.S. went into a frenzy of overblown responses, leading to the criminal launching of the Iraq war. Somewhere in the wake of that reaction, the Bush-Cheney administration announced that it was radically increasing research on potential pandemic pathogens, with the research having dual use against natural and man-made pathogens. The 2005 public announcement of the pandemic preparedness plan was remarkably thoughtful, laying out many important steps that we are still failing to implement. The scientist Anthony Fauci was put in charge of the research, rather than the military. I thought, and probably mentioned to a few friends, that this was at least a thin silver lining, something useful against a major natural danger that had been neglected.
I was mistaken.
What Next
We agree that most scientific research should be defended against the massive attacks from the Trump administration. We agree that the part of that research that involves playing with potentially risky new pathogens should stop. It’s not especially necessary to persuade major governments of the danger. They’re now well aware of it and some seem to have been well aware since early 2020. They’re going to do whatever they think is in their best interests, e.g. maybe move the riskiest research to military labs. It’s not necessary to persuade most of the US public of the danger since an overwhelming majority already think Covid came from a lab, often with some weird conspiracy twists added.
So why do I care about getting the scientific answer right? I like to think it’s a lifelong passion for scientific truth, but diagnosing one’s own motivations is a fool’s errand. Empirically, it’s from the same compulsion that has led me to help catch a couple of fake pollsters and to publish many correct Comments on both purely technical physics papers and on some physics education research papers with an ideological edge. But there’s also a political reason.
The people who need to hear something like the straight story are the liberal-leaning 17% or so of the American public who think that the lab leak idea is improbable, including the 5% of the public, again concentrated among liberals, who are quite sure it didn’t happen. Many of those think that lab leak is a far-fetched right-wing fable. Over the years you’ve made a point of challenging the mild liberal consensus, sometimes to an extent that you seem to now regret. The origin of Covid is an odd topic on which to choose to coddle the comforting canonical viewpoint. Beliefs about covid origins are hardly the hottest-button topic in politics but it is among the topics for which encouraging liberals to stay in their bubble isolates them from the large majority and gives the impression of willful lack of realism. It’s particularly hard to argue against the actual conspiracy theories, often tied in to anti-vax fables, while holding on to the now implausible zoonotic account. Why should anyone outside the choir listen?
Since the Covid origins question is not as personally charged as some other issues, it might be a good one to start loosening up the group-think that has politically weakened the nominal left. As a tiny, tiny first step, it would help to stop genuflecting to the most clearly false claims on this scientific topic. P2022 is flat-out wrong. W2022 dissolves under inspection by standard techniques. Your big Bayes factor just ain’t there. What’s left of your analysis strongly favors lab leak. Perhaps there’s no point in saying so, since reaching that conclusion with much confidence would require another big investment of time. It’s a disservice to your followers, however, not to at least say “whoops, scratch that, I meant maybe.”
Maybe it's a cheap point to make, but the CIA, the FBI, and intelligence agencies writ large aren't largely comprised of rebels, self-styled or otherwise.
There are intel lab leakers, data-oriented lab leakers, academic ones, activist ones, crazy ones, bat-shit crazy ones, MAGA ones, and Jamie Metzl. Conversely, the other side is a small academic niche and the rationalist autists they've persuaded.
Professor Fouchier went on national TV saying he heard of an outbreak in Wuhan in the first week of December.
That means either Professor Fouchier was lying on national TV, with no absolutely no incentive to lie, or the Chinese government was lying to the WHO about the timing of the first case on 8th December, with a gigantic incentive to lie as they wanted to hide how long they had been covering it up in breach of international treaty obligations, and appear highly competent by discovering a new flu-like virus within 3 weeks of emergence in the middle of the flu season, which seems objectively impossible.
I'm not sure which way round the Bayes factor for this would be, can anyone help?
The entire market origin theory rests on the case dataset published by China, so it would be a shame if it turned out to be about as accurate as the reported number of deaths, which was at least 20 times lower than any plausible number.