
An Inconvenient Probability
A robust Bayesian look at Covid origins

The latest version is here. These older version are left up to avoid hiding any old mistakes etc.
Introduction
Early on in the Covid pandemic, I took a preliminary look at the relative probabilities that SARS-CoV-2 (SC2) came from some sort of lab leak vs. more traditional direct zoonotic paths. Although zoonotic origins have been more common historically, the start in Wuhan where the suspect lab work was concentrated left the two possibilities with comparable probabilities. That result seemed convenient because it left strong motivation both to increase surveillance against future zoonosis and to stringently regulate dangerous lab work. Since then there have been major changes in circumstances that change both the balance of the evidence and the balance of the consequences of looking at the evidence. I think these warrant taking another look.
The origins discourse has become increasingly polarized with different opinions often tied to a package of other views. To avoid having some readers turn away out of aversion to some views that have become associated with the suspicion of a lab leak, it may help to first clarify why I think the question is important and to name some of the claims that I’m not making before plunging into the more specific analysis of probabilities.
I’m now more concerned about dangerous work being accelerated rather than useful work being over-regulated. This is not a specifically Chinese problem. Work with dangerous new viruses is planned or underway in Madison, and the Netherlands, with some questionable work in Boston. I’m not suggesting that the US or other western governments should officially say that they think SC2 started from a Wuhan lab. That would make it harder to work with China on the crucial issues of global warming and even on international pathogen safety regulation itself. (I just signed a letter “urging renewal of US-China Protocol on Scientific and Technological Cooperation”.) I’m definitely not endorsing the cruel opposition to strenuous public health measures that seems to have become associated with skepticism about the zoonotic account.
In what follows I will try to objectively calculate the odds that SC2 came from a lab, in the hope that will be useful to anyone thinking about future research policy. The underlying motivation for this effort has been eloquently described by David Relman, who has also provided a nice non-quantitative outline of the general types of evidence supporting different SC2 origins hypotheses.
The method used may also help readers to evaluate other important issues without relying too much on group loyalties. The method is explicitly ready for correction based on improved reasoning or new evidence. [9/9/23: updates are coming soon thanks to comments from people on both sides of the question.] Our method will be robust Bayesian analysis, a systematic way of updating beliefs without putting too much weight on either one’s prior beliefs or the new evidence. Bayesian analysis does not insist that any single piece of evidence be “dispositive” or fit into any rigid qualitative verbal category. Some hypotheses can start with a big subjective head-start, but none are granted categorical qualitative superiority as “null hypotheses” and none have to carry a qualitatively distinct “burden of proof”. Each piece of evidence gets some quantitative weight based on its consistency with competing hypotheses.
In practice there are subjective judgments not only about the prior probabilities of different hypotheses but also about the proper weights to place on different pieces of evidence. “Robust Bayesian analysis“ provides a systematic way of taking those uncertainties about the evidence into account. I will use a simple version of such robust Bayesian analysis.
I am aware of six more-or-less published Bayesian analyses of SC2 origins, other than my own very preliminary inconclusive one. The four that attempt to be comprehensive come to conclusions similar to the one I shall reach here. These are discussed in Appendix 1.
I will focus on comparing the probability that SC2 originated in wildlife vs. the probability that it originated in work similar to that described in the 2018 DEFUSE grant proposal submitted to the US Defense Advanced Research Project Agency from institutions that included the University of North Carolina (UNC), the National University of Singapore, and the EcoHealth Alliance as well as the Wuhan Institute of Virology (WIV). (For brevity I’ll just refer to this proposal as DEFUSE.) Although DEFUSE was not funded by DARPA, anyone who has run a grant-supported research lab knows that work on yet-to-be-funded projects routinely continues except when it requires major new expenses such as purchasing large equipment items.
I will not discuss any claims about bioweapons research. It is not exactly likely that a secret military project would request funding from DARPA for work shared between UNC and WIV.
My analysis will not make use of the rumors of roadblocks around WIV, cell-phone use gaps, sick WIV researchers, disappearances of researchers, etc. That sort of evidence might someday be important but at this point I can’t sort it out from the haze of politically motivated reports. Mumbled inconclusive evidence-free executive summaries from various agencies are even less useful. The biological and geographic data are much more suited to reliable analysis.
The main technical portions will be unpleasantly long-winded since for a highly contentious question it’s necessary to supply supporting arguments. Although parts may look abstract to non-mathematical readers, all the arguments will be accessible and transparent, in contrast to the opaque complex modeling used in some well-known papers. For the key scientific points I will provide standard references. At some points I bolster some arguments with vivid quotes from key advocates of the zoonotic hypothesis, providing convenient links to secondary sources. The quotes may also be obtained from searchable .pdf’s of slack and email correspondence.
The outline is to
1. Give a short non-technical preview.
2. Introduce the robust Bayesian method of estimating probabilities, along with some notation.
3. Discuss a reasonable rough consensus starting point for the estimation, i.e. the “priors” with an update based on the pandemic starting in Wuhan.
4. Discuss whether the main papers that have claimed to demonstrate a zoonotic origin via the wildlife trade should lead us to update our odds estimate.
5. Update the odds estimate using a variety of other evidence.
6. Present brief thoughts about implications for future actions.
Preview
I will denote three general competitive hypotheses:
ZW: zoonotic source transmitted via wildlife to people, suspected via a wet-market.
ZL: zoonotic source transmitted to people via lab activities sampling, transporting or otherwise handling viruses.
LL: a laboratory-modified source, leaked in some lab mishap.
The viral signatures of ZW and ZL would be similar, so the ratio of their probabilities would be estimated from knowledge of intermediate wildlife hosts, of the lab practices in handling viral samples, and detailed locations of initial cases. Demaneuf and De Maistre wrote up a Bayesian discussion of that issue in 2020, concluding that the probability of ZW, i.e. P(ZW), and the probability of ZL, i.e. P(ZL), were about equal. Much of their analysis, particularly of prior probabilities, is close to the arguments I use here, but written more gracefully and with more thorough documentation. They use a different way of accounting for uncertainties than I do, but unlike some other estimates their method is transparent and rational. Nevertheless, here I’ll just compare the probability P(ZW) to that of the LL lab account, P(LL), because sequence data point to a lab involvement in generating the viral sequence, so that P(ZL) will itself be smaller than P(LL).
Ratios of probabilities such as P(LL)/P(ZW) are called odds. It’s easier to think in terms of odds for most of the argument because the rule for updating odds to take into account new evidence is a bit simpler than the rule for updating probabilities.
I’ll start with odds that heavily favor ZW, historically the common origin of most new epidemics. Then I’ll update using several important facts. The most immediately obvious is that the location of the initial outbreak was Wuhan, the location of a major research lab that had submitted a grant proposal that included modifying bat coronaviruses in the way later found in SC2 and that had already been noted for difficulties with lab safety. That location could also have occurred by accidental coincidence for ZW, but we shall see that it’s not hard to approximately convert the coincidence to a factor objectively increasing the odds of LL. Here’s a beginning non-technical explanation of how the odds get updated.
I’ll start with a consensus view, that the prior guess would be that P(LL) is much less than P(ZW). That corresponds to the standard idea that you would call ZW the null hypothesis, i.e. the boring first guess. Rather than treat the null as qualitatively sacred I’ll just leave it as initially quantitatively more probable by a crudely estimated factor.
Now we get to the simple part that has often been either dismissed or over-emphasized. Both P(ZW) and P(LL) come from sums of tiny probabilities for each individual person. P(LL) comes mostly from a sum over individuals in Wuhan. P(ZW) comes from a sum over a much larger set of individuals spread over China and southeast Asia. Since we know with confidence that this pandemic started in Wuhan, restricting the sum of individual probabilities to people around Wuhan doesn’t change P(LL) much but eliminates most of the contributions to P(ZW). Wuhan has less than 1% of China’s population. That means we need to increase whatever P(LL)/P(ZW) odds we started with by about a factor of 100, since the denominator is reduced.
Further updates following the same logic come from other data. The most important single update will come from a special genetic sequence that codes for the furin cleavage site (FCS) where the UNC-WIV-EHA DEFUSE proposal suggested adding a tiny piece of protein sequence to a natural coronavirus sequence. The tiny extra part of SC2’s spike protein, the FCS that is absent in its wild relatives, has nucleotide coding that is almost never found in other parts of SC2 or in its relatives but is typical for the most relevant known designed sequences– e.g. the mRNA vaccines. One can picture this via a map representing RNA sequences, in which SC2 appeared at a location that is almost uninhabited naturally but where a bustling city of lab sequences resides. This coincidence turns out to be even more numerically striking than for the physical location.
Once these two simple updates are included we’ll have P(LL) much larger than P(ZW) even if we start with a generously high but plausible preference for ZW. A look at other updates, both ones claimed to favor ZW and some of the ones claimed to favor LL, will swing the odds further toward LL. P(ZW) will shrink to about 1%, and is saved from shrinking much further only by allowance for uncertainties.
This openly crude and approximate form of argument may alarm readers who are not accustomed to the Fermi-style calculations routinely used by physicists. In this sort of calculation one doesn’t worry much about minor distinctions between similar factors, e.g. 8 and 12, because the arguments are not generally that precise. Sometimes the large uncertainties in such a calculation render the conclusion useless, but this turns out not to be one of those cases.
Methods
The standard logical procedure to calculate the odds, P(LL)/P(ZW), is to combine some rough prior sense of the odds with judgments of how consistent new pieces of evidence are with the LL and ZW hypotheses. Bayes’ Theorem provides the rule for how to do this. (See e.g. this introduction.)
One starts with some roughly estimated odds based on prior knowledge:
P0(LL)/P0(ZW). Then one updates the odds based on new observations. The probabilities that you would see those observations if the hypothesis (LL or ZW) were true are denoted P(observations|LL) and P(observations|ZW), called the “likelihoods” of LL and ZW. Assuming these likelihoods are themselves known, Bayes’ Theorem tells us the new “posterior” odds are
P(LL)/P(ZW) = (P0(LL)/P0(ZW))*(P(observations|LL)/P(observations|ZW)).
In practice, it’s hard to reason about all the observations lumped together, so we break them up into more or less independent pieces and do the odds update using the product of the likelihood ratios for those pieces.
P(LL)/P(ZW) = (P0(LL)/P0(ZW) )*(P(obs1|LL)/P(obs1|ZW))*(P(obs2|LL)/P(obs2|ZW))…… *(P(obsn|LL)/P(obsn|ZW))
At this point it’s necessary to recognize that not only the prior odds P0(LL)/P0(ZW) but also the likelihoods involve some subjective estimates. In order to obtain a convincing answer we need to include some range of plausible values for each likelihood ratio, i.e. use robust Bayesian methods. As we shall see, inclusion of the uncertainties is important because realistic recognition of the uncertainties will tend to pull the final odds back from an extreme value towards one.
Once our odds become products of factors of which more than one have some range of possible values, our expected value for the product is no longer equal to the product of the expected values. Since the expected value of a sum is just the sum of the expected values it’s convenient to convert the product to a sum by taking the logarithms of all the factors.
ln(P(LL)/P(ZW)) = ln(P0(LL)/P0(ZW))+ln(P(obs1|LL)/P(obs1|ZW)) … +ln(P(obsn|LL)/P(obsn|ZW)) = logit0 + logit1 … +logitN
where “logit” is used for brevity.
At each stage I will include a crude estimate of the uncertainty in the estimate of each factor, expressed as an estimated standard error of its logit. The final odds estimate will be obtained from a logit distribution centered on the sum of the logits with a width determined by the square root of the sum of the squares of the standard errors, since the errors in different factors are presumed to be independent of each other. A further approximation, treating the net logit distribution as Gaussian, will then allow us to calculate net odds taking the uncertainties of the factors into account.
Along the way we shall see several observed features that perhaps should give important likelihood factors but for which there’s substantial uncertainty. I will not omit any that I think would favor ZW but will drop some that I think tend to favor LL. The peculiarity of some features under ZW will not be used to penalize ZW’s odds if those features are likely to have a notable selective advantage, since we are only viewing the virus after an evolution that can selectively amplify peculiar accidents. I will include some small factors when the sign of their logit is unambiguous, e.g. a factor from the lack of any detection of a wildlife host.
The quantitative arguments
Prior odds
Let’s start with the fuzzy prior odds. In my lifetime, starting in 1949, there have been seven other significant (>10k dead) worldwide pandemics. Although, as the book Pandora’s Gamble amply documents, pathogen lab leaks are common, including in the US, they are almost always caught before the diseases spread. Nevertheless, at least one pandemic (1977 H1N1) came from some accident in handling viral material. So if we just wanted to base our priors on that, we’d say, very crudely,
(P0(LL or ZL)/P0(ZW) ) = 1/7 = ~0.1.
There’s an important caveat, however. So far as we know, all of the past epidemics that came from labs (e.g. 1967 Marburg viral disease in Europe, 1979 anthrax in Sverdlovsk, 1977 influenza A/H1N1) were caused by natural pathogens. That’s not surprising, since until recently nobody was doing much pathogen modification in labs. The main modern method was only patented in 2006. Without lab modification, only ZW and ZL would be viable hypotheses.
We know, however, that lots of modifications are underway now in many labs. The dangers were perceived as substantial enough for the Obama administration to at least nominally ban funding research involving dangerous gain-of-function modifications of pathogens. When that ban was lifted under Trump in 2017, Marc Lipsitch and Carl Bergstrom raised alarms. Lipsitch wrote: “ [I] worry that human error could lead to the accidental release of a virus that has been enhanced in the lab so that it is more deadly or more contagious than it already is. There have already been accidents involving pathogens. For example, in 2014, dozens of workers at a U.S. Centers for Disease Control and Prevention lab were accidentally exposed to anthrax that was improperly handled.” Bergstrom tweeted a similar warning. It is hard to see how such warnings would make sense if expert opinion held that the recent probability of a dangerous lab leak of a novel virus was negligible. For at least the last decade the prior probability P0(LL) of escape of a modified pathogen has not been negligible.
Several papers have been published on lab-modified viruses, e.g. one that demonstrated potential for modified bat coronaviruses to become dangerous to humans: “Using the SARS-CoV reverse genetics system, we generated and characterized a chimeric virus expressing the spike of bat coronavirus SHC014 in a mouse-adapted SARS-CoV backbone.” At least one paper specifically described adding an FCS to a SARS-CoV virus. The 2018 DEFUSE proposal from WIV included plans for just such modifications of coronaviruses. Even K. G. Andersen, the lead author of the first key paper (“Proximal Origins”) claiming to show that LL was implausible, initially thought “…that the lab escape version of this is so friggin’ likely to have happened because they were already doing this type of work and the molecular data is fully consistent with that scenario.” That view is inconsistent with claims that the prior P0(LL) was extremely small, although it neither quantifies “friggin’ likely” nor establishes how much of “friggin’ likely” would be attributed to priors and how much to molecular data whose analysis may have since changed.
Should our prior probability of a pandemic from a new lab virus be raised or lowered compared to our old empirical probability of a lab-origin pandemic in light of the new prevalence of modern research in which pathogens are modified? On the one hand, only some of the viruses studied in labs are new, so the probability that a leak would be of something new is less than the net probability of any leak. On the other hand, more lab work is being done than in the past, raising the overall leak probability. Furthermore, we are interested only in the probability of a major pandemic-causing leak, and that is going to be higher for new viruses than for old ones since there’s some population immunity for the old ones.
I think the prior odds P0(LL or ZL)/P0(ZW) should be at least about the same as the old empirical ~0.1, but want to be conservative here in order not to lose reasonable readers who disagree before we get to the core evidence. So let’s just make a crude but quite conservative estimate for starters:
P0(LL)/P0(ZW) = ~0.01.
We shall see that it is in the range others consider reasonable. (It is at the upper end of J. Seymour’s range, but he provides no rationale for his choices, which seem incompatible with the historical record.)
Although each subsequent likelihood ratio adjustment has its own uncertainty, the uncertainty of these prior odds will be the most important one. Let’s estimate the uncertainty in the prior odds as about a factor of 10 either way. It will be convenient when we put together the pieces to also describe factors and their uncertainties in terms of the natural logs of the odds, i.e. logits. Our prior is then equivalent to
logit0 = -4.6 ±2.3
where the ±2.3, equivalent to the factor of 10, is meant to roughly show the standard error in estimating the logit. A standard error of 2.3 allows and even requires that errors outside the ±2.3 range are possible, although not very probable. “4.6” is not meant to convey false precision, just to translate a rough estimate (x100) into convenient units.
We’ll return to check how well that prior agrees with expert opinion after updating to include knowing that the pandemic started in Wuhan. The reason is that some expert opinions expressed after the pandemic started already integrated the priors with that knowledge.
Starting in Wuhan
Now let’s take the first, most obvious piece of evidence—the pandemic started in Wuhan. Even without any formal Bayesian notation it’s easy to understand why that shifts the odds heavily toward LL or ZL. The probability of ZW comes from a sum over people and wet markets spread over China, Laos, etc.. A recent paper arguing entirely within the ZW framework has shown that SC2 is a fairly recent chimera of known relatives living in or near southern Yunnan, and that transmission via bats is essentially local on the relevant time scale. Wuhan is sufficiently remote from those locations that WIV has used Wuhan residents as negative controls for the presence of antibodies to SARS-related viruses. Thus Wuhan residents are not particularly likely to pick up infections of this sort from wildlife. About 0.7% of the population of China lives in Wuhan.
Wuhan has fewer wet markets than expected from its population. Wuhan had only 17 wet market shops in four markets. Overall, China has about 44,000 wet markets according to one source and about 4600 according to another. I do not know the reason for the discrepancy, perhaps a count of shops vs. markets, but even using the most extreme numbers Wuhan would have less than 0.4% of the wet markets, probably less than 0.1%. So knowing that the pandemic started in Wuhan, of all places, gives:
P(Wuhan|ZW) < 0.01.
What is P(Wuhan|LL)? We know that WIV’s DEFUSE specifically described planned coronavirus modifications including FCS insertion, modification of a feature called an N-linked glycan, and major modifications of the receptor binding domain compared to natural strains, all features later found in SC2. We know there were U.S. State Department cables warning specifically that bat coronavirus work in Wuhan faced safety challenges. We know that the DEFUSE proposal claimed WIV had more than 180 relevant coronavirus sequences, apparently including many unpublished ones. Although there are undoubtedly other cities where some coronavirus work is going on, if someone with this prior knowledge heard that a lab leak had started a pandemic of a coronavirus with an FCS etc., they would have been pretty sure that the location was Wuhan:
P(Wuhan|LL, FCS,…) is not a lot less than 1.
This first likelihood ratio, from Wuhan being the starting location, is then:
P(Wuhan|LL)/P(Wuhan|ZW) = ~100
We can estimate the standard error in that estimate as about a factor of 2.
Our logit estimate will then be updated by
logit1 = +4.6 ±0.7.
Again, the “4.6” is not meant to convey false precision.
Priors adjusted for Wuhan start
At this point of the analysis the combined logit is ~0, i.e. the chances are about equal. Let’s check that our odds are reasonable at this point, based on the combination of priors and that the outbreak started in Wuhan. Demaneuf and De Maistre looked in detail at past evidence for various scenarios of natural and lab-related outbreaks. Without considering sequence features beyond that the virus is SARS-related they conservatively estimate that the lab-related to non-lab-related odds are about one-to-one for an outbreak in Wuhan, again in agreement with the odds we use.
Now let’s double-check our starting point that the chances for ZW and LL in Wuhan were comparable. One serious pre-Covid paper estimated the chance of a human transmissible leak at 0.3%/year for each lab. Another careful pre-Covid analysis of experiences of labs using good but not extreme biosafety practices (“BSL3”) estimated that the yearly chance of a major human-transmissable leak was in the range of 0.01% to 0.1% per lab. For a large lab doing much of its work at a much lower safety level (BSL2) the chances would be higher, easily >0.1%/year. According to the lead coronavirus researcher at WIV, Shi Zhengli, “coronavirus research in our laboratory is conducted in BSL-2 or BSL-3 laboratories.“ For comparison, newly important zoonotic diseases have been identified in China at a rate of about 0.4/year. With Wuhan having about 0.7% of China’s population, not located near coronavirus hotspots, its local rate should be less than 0.3%/year. Once again, we have roughly even odds for a lab origin or a natural origin for a new pandemic starting in Wuhan.
Finally, let’s triple-check by looking at the impressions of the lead author of Proximal Origins. Andersen wrote his colleagues on 2/2/2020 “Natural selection and accidental release are both plausible scenarios explaining the data - and a priori should be equally weighed as possible explanations. The presence of furin a posteriori moves me slightly more towards accidental release, …” Based on general priors plus knowledge of the Wuhan origin and before looking at more detailed data such as the FCS, Andersen thought the probabilities were about equal, which is just the result we have reached at the same point.
The key papers arguing for zoonosis
Proximal Origins
Now let’s look at the three main papers on which claims that the evidence points to ZW rest. The first is the Proximal Origins paper, whose valid point was that ZW was at least possible. Its initially submitted version concluded logically that therefore other accounts were “not necessary”. That conclusion is implicit in all the Bayesian analyses, which neither assume nor conclude that P(ZW)=0.
The final version of Proximal Origins changed that conclusion under pressure from the journal to the illogical claim that therefore accounts other than ZW were “implausible”. To the extent that the paper had an argument for LL being implausible it was based on the assumptions that a lab would pick a computationally estimated maximally human-specialized receptor binding domain rather than just a very well-adapted human receptor binding domain and that seamless modern methods of sequence modifications would not have been used. Neither assumption made sense, invalidating the conclusion. Defense Department analysts Chretien and Cutlip already noted in May 2020: “The arguments that Andersen et al. use to support a natural-origin scenario for SARS CoV-2 are not based on scientific analysis, but on unwarranted assumptions.” The later release of the DEFUSE proposal further clarified that the sorts of lab modifications that Proximal Origins argued against were not the sort that WIV had been planning. Thus Proximal Origins contains nothing that would lead us to update our odds in either direction.
As further confirmation, we now know that even weeks after Proximal Origins was published its lead author did not have confidence in its conclusions or even believe its key arguments. On 4/16/2020 Andersen wrote his coauthors : “I'm still not fully convinced that no culture was involved. If culture was involved, then the prior completely changes …What concerns me here are some of the comments by Shi in the SciAm article (“I had to check the lab”, etc.) and the fact that the furin site is being messed with in vitro. … no obvious signs of engineering anywhere, but that furin site could still have been inserted via gibson assembly (and clearly creating the reverse genetic system isn't hard -the Germans managed to do exactly that for SARS-CoV-2 in less than a month.”
Phylogeny and location: Pekar et al. and Worobey et al.
The next papers involve phylogenetic data and location data. Readers should be forewarned that the likelihood factors for their combination do not factorize into separate contributions. The reason is that the locations data were used to support one particular version of the ZW hypothesis and the phylogenetic data make that particular version implausible although on their own they would say little to disfavor the general ZW hypothesis.
Pekar et al. argued based on computer simulations of a simplified model of how the infection would spread that the presence of two lineages (A and B) differing by two point mutations in the nucleic acid sequence without intermediate cases was unlikely if all human cases descended from a single most recent common ancestor (MRCA) that was in some human. They claimed to obtain Bayesian odds of ~60 favoring a picture in which the MRCA was in another animal shortly before two separate spillovers to humans. There is no obvious reason why having an MRCA in some other animal a few transmission cycles before two spillovers to humans would say much about whether the other animal was a standard humanized mouse in a lab or an unspecified wildlife animal in a market. For example, multiple workers were exposed to Marburg fever in the lab and the Sverdlovsk anthrax cases included multiple strains. Further discussion of the Pekar et al. model seems irrelevant to our question, but I’ll include a brief discussion in Appendix 2 about some of the major technical problems of the paper.
Let’s step back from complicated, assumption-laden modeling that seems irrelevant to our ZW vs. LL comparison to look at what the lineage data seem to say prima facie. (Jesse Bloom and Trevor Bedford wrote a convenient introductory discussion.) Lineage A shares with related natural viruses the two nucleotides that differ from B. Thus lineage A was the better candidate for being ancestral, as Pekar et al. acknowledged. Pekar et al. describe 23 distinct reversions out of 654 distinct substitutions in the early evolution of SC2. The chance that when two lineages are separated by two mutations (2 nucleotides, “2nt”) both those mutations would be reversions is then roughly (23/654)2 = 0.00124 = ~1/800. At this point that conclusion tells us nothing about P(LL)/P(ZW), but it will become important when integrated with information about locations of early cases and early viral traces.
Lineage A was almost entirely absent from the main suspected site of the wildlife spillover, the Huanan Seafood Market (HSM). Although many traces of B were found in HSM, traces of A were found only on one glove, with additional mutations indicating that it was not from an early case. Thus the sequence data indicate that lineage A was quite unlikely to have originated at HSM. This conclusion applies whether or not the spillover that led to lineage A was the only one or whether there was a separate spillover to lineage B.
More complete data and analysis indicate that neither A nor B was the MRCA. The MRCA seems to have differed from lineage B by 3nt shared with wild relatives, not 2nt. The MRCA was probably present in Oct. 2019, with the first spillover case likely to have occurred weeks earlier. Bloom has taken a deeper look at the lineage data finding more early sequences and confirming that the MRCA differed from B by 3nt. Bloom finds more early lineage A and other sequences closer to the MRCA at multiple locations away from the market, including other parts of Wuhan, other parts of China, and other countries. The phylogeny data thus seem inconsistent with HSM being the only spillover site.
At this point it is tempting to add a Bayes factor disfavoring ZW, since the phylogenetic results seem incompatible with an exclusively HSM spillover. Nevertheless, ZW spillovers at other locations would be possible. The phylogenetic data do play a key role in strongly disfavoring a particular version of ZW in which the spillover or spillovers to humans occurred at HSM.
We’ve looked at whether the sequences found in the HSM were reasonably compatible with that being the first spillover site (they weren’t) but we haven’t made the equivalent test for WIV. Depending on what sequences were there, one could end up with a Bayes factor either favoring ZW or LL. Unfortunately we have little information. In Sept. 2019 WIV removed public access to its sequence collection. Publication of newly gathered sequences seems to have abruptly stopped with those gathered in 2016, at least according to the data I’ve been provided. (If someone knows of updates that would be helpful.) Y. Deigin discusses further omissions from public disclosure of what sequences were known as well as of when and where they were obtained.
Some people have read this missing-data situation as indicating a systematic cover-up of some embarrassing sequence data. Others consider the lack of evidence for a close match of a WIV sequence to SC2 as indicating that SC2 was unlikely to come from WIV. Others have said it’s just reflexive bureaucratic secrecy with no particular implications. An evaluation of the likelihoods under ZW, ZL, and LL of the removals of various sorts of data from Wuhan and the inconsistencies between various statements of prominent virologists would be an interesting project for a social scientist, but not one I will use to update here.
In combining the lineage and case location data we can simplify a bit by using one point on which there is unanimity– if there were more than one spillover either all or none were lab-related. Is there evidence that lineage B spilled over to humans at HSM? If so, that would support ZW.
The widely publicized paper by Worobey et al. used case location data to argue that HSM was not just a superspreading location but also the location of the spillover to humans. Worobey et al. argue that since there were hundreds of plausible superspreading locations it would require a remarkable coincidence, with probability ~1/400, for a possible spillover site, HSM, to be the first ascertained spreading site unless it were the actual spillover site. One can get a preliminary empirical feel for how much of a coincidence that would be by looking at the first notable ascertained outbreak in Beijing some 56 days after initial cases were controlled. It occurred at the Xinfadi wet market, which could not have been the site of the months-earlier spillover. Apparently first ascertainment of spread of a pre-existing human virus is not so unlikely to be located at a wet market.
The case data Worobey et al. used omitted about 35% of the known cases for unspecified reasons, a crucial problem for an analysis based on spatial correlations. Proximal Origins author Ian Lipkin described the Worobey et al. analysis as "… based on unverifiable data sets…" The collection of known cases already was biased because proximity and ties to HSM were used as criteria for detecting cases in the first place. A report from the WHO and the Chinese CDC looking at the case location data concluded “Many of the early cases were associated with the Huanan market, but a similar number of cases were associated with other markets and some were not associated with any markets….No firm conclusion therefore about the role of the Huanan Market can be drawn.”
Worobey et al. include a map of locations of requests to the Weibo web site for assistance with Covid-like disease, which provides a way of looking at the location distribution within Wuhan without selective omission of cases. The earliest map presented shows a tight cluster near to but not centered on HSM. Instead it clusters tightly more than 3 km southeast on a Wuhan CDC site (not part of WIV) where BSL2 viral work was done. Just before the time of the first officially recorded cases the CDC opened a new site within 300m of HSM, indistinguishable from the HSM site via the sorts of data used in Worobey et al. Thus how important HSM was even as a later superspreading site is unclear.
Worobey et al. present another argument— that the distribution of SC2 RNA within HSM pointed to a spillover from some wildlife there. If correct, that argument would be more directly relevant to whether a spillover occurred at HSM than are the locations of cases after Covid became more widespread.
The positive SC2 RNA reads did tend to cluster in the general vicinity of some of the HSM wildlife stalls, even after correcting for the biased sampling that focused on that area. That area, however, is also where bathrooms and a Mah Jong room are located, both likely spreading sites. A finer-grained map using the Worobey data showed the hot spot to be centered on the bathroom/Mah Jong spot, not the nearby wildlife stalls.
In a short-lived coda, there were many press stories that SC2 RNA found in a stall with DNA of a raccoon dog showed that species to be the intermediate host. The presence of wildlife in the market was not news– it is implicit already in our priors. The question was whether there was some particular connection between that wildlife and SC2. When Bloom went over the actual data for the individual samples, he found that particular sample had almost undetectable SC2 RNA, far less than many others. Overall, sample-by-sample SC2 RNA correlated negatively with the presence of DNA from possible non-human hosts.
Thus the internal SC2 RNA data make it unlikely that wildlife had any direct connection with SC2 spread in HSM. As the head of China’s CDC concluded, “At first, we assumed the seafood market might have the virus, but now the market is more like a victim. The novel coronavirus had existed long before”. Nonetheless, to be conservative I will not include a Bayes factor disfavoring the general ZW hypothesis at this point.
Intermediate hosts
The failure to find any positive statistical association of SC2 RNA with any plausible intermediate host in the HSM points to a larger issue. For both the important recently spilled-over human coronaviruses, SARS-CoV-1 and MERS, intermediate wildlife hosts were found. In contrast, no wildlife intermediary has been found anywhere for SC2 despite intense searches. According to the Lancet Commission “Despite the testing of more than 80000 samples from a range of wild and farm animal species in China collected between 2015 and March, 2020, no cases of SARS-CoV-2 infection have been identified.”
Intermediate hosts were found for 3 of the 4 other recently identified human betacoronaviruses, with the missing one (HCoV-HKU1) causing a relatively minor disease that provoked relatively little attention. A broader review of human coronaviruses finds that intermediate hosts have been identified for 7 of the 9 described, not counting SC2.
Given the enormous attention paid to SC2, I think the probability of not finding any intermediate under the ZW hypothesis would be less than for the other coronaviruses, but we can conservatively estimate the logarithm of probabilities consistent with the observations for the other coronaviruses. I calculate the expected value of ln(P(no wildlife host found|ZW)) assuming a uniform prior on the probability of non-observation. (See Appendix 3) Although the identification of intermediate hosts for the two most relevant cases produces the most negative expected
lnP(no wildlife host found|ZW) it has large uncertainty due to the very small sample. The larger samples give less negative values for lnP(no wildlife host found|ZW) but with reduced uncertainty. (See Appendix 3)
Of course, P(no wildlife host|LL) =1. Thus based on the absence of any intermediate host samples expected for ZW our probabilities should be updated by a modest likelihood ratio of ~4, corresponding to:
Logit2 = 1.4 ±0.6.
To be symmetrical, one should also consider whether there are any traces of an intermediate host of the type that might be found under the LL hypothesis, i.e. either cell cultures or humanized mice that would be used in the type of work proposed in DEFUSE. SC2 sequences did show up in data from the Sangon sequencing lab in samples contaminated with standard lab Vero culture cells, the same type of lab culture cells that DEFUSE had described planning to use. Three mutations that differ from the initial SC2 sequence but are shared with related wild viruses were detected in these samples, although with poor data quality. Thus there is a plausible but not proven sample with an intermediate ancestral host consistent with LL. Since human sequences are mixed in with these samples and their date is not firmly established, they are also reasonably consistent with ZW. Thus although some have cited the Sangon observation as strong evidence for LL it doesn’t let us update the odds with much confidence. [9/9/23: I’ve since realized that those three mutations are not just generic ancestral but apparently specific matches to the MRCA sequence estimated by Kumar et al. I’m trying to find some excuse to only modestly update the odds rather than switch into the smoking-gun mode. ]
The FCS
Some LL advocates have argued that the mere fact that SC2 has an FCS is strong evidence for LL since no close relative of SC2 has an FCS and DEFUSE proposed adding an FCS. As we have seen, even the lead author of Proximal Origins thought the FCS was at least some evidence favoring LL. Nevertheless, the argument that having an FCS gives a major factor is exaggerated, since it would only apply to some generic randomly picked relative. SC2 is not randomly picked. We are only discussing SC2 because it caused a pandemic. So far as we know having an FCS may be common in the subset of hypothetical related viruses that are capable of causing a pandemic. In other words P(FCS|ZW, pandemic) may be nearly 1 even though P(FCS|ZW) is much less than 1 for some generic sarbecovirus. Therefore I will not use the mere existence of an FCS to update the odds. (See Appendix 4 for a consolidated discussion of how the FCS data are used here.)
The specific contents of the FCS, however, do provide strong evidence. Focusing on the internal details of the FCS site is not cherry-picking statistical oddities from a large range of possibilities, since it is specifically the tiny FCS insertion that seems so peculiar for this type of virus and so predictable for DEFUSE-style synthesis. The details of the FCS initially struck David Baltimore as a smoking gun for LL, although he later moderated that claim. As we saw in our introduction of the methods, rather than categorizing each such unusual feature as either a smoking gun or mere coincidence, Bayesian analysis assigns each feature a quantitative odds update factor. Events that are unusual under some hypothesis do not rule out that hypothesis but they do constitute evidence against it if the events are more likely under a competing hypothesis.
The SC2 FCS has two adjacent arginines (Arg’s), each coded for by the nucleotide codon CGG. CGG is the least common of the 6 Arg codons in all related natural viruses. CGG is only used for ~2.6% of the Arg’s in the rest of SC2. None of the other 40 Arg’s on the spike protein use CGG. There’s no reason to think that the two codon choices would correlated under the ZW hypothesis, so we can treat them as approximately independent, leaving P(CGGCGG|ZW)= 0.0262 = ~0.0007.
One can check the independence assumption using Arg pairs in related viruses: “…we have checked all 255 sarbecovirus strains present in GenBank that have protein annotations, and with the exception of the SARS-CoV-2 FCS, none have two consecutive arginines coded by CGGCGG anywhere in their genomes (on average, each sarbecovirus strain has 12 arginine doublets in its annotated proteins)”. With zero of more than 3000 other Arg pairs coded CGGCGG, our estimate P(CGGCGG|ZW) = ~1/1400 is conservative. The occasional claims that a sequence that happened to create an effective FCS in humans could have been inserted at the S1/S2 junction by accidental copy error in an ancestral virus are irrelevant to this estimate, since the potential insertion sources are subject to the same codon probabilities.
We need to compare that with an estimate of P(CGGCGG|LL). [9/9/23: My following discussion of that is somewhat naive and will be modified, I think slightly reducing the odds.] CGG is actually rather common outside the coronavirus family, coding for 20% of human Arg. When sequences are synthesized for use in hosts, they are typically “codon optimized”, using the more common host codons, such as CGG in humans, even more frequently than they are found in the host. Thus a minimum estimate of P(CGGCGG|LL) would be 0.22=0.04. We need to look at some synthesized sequences to get a better estimate.
I found two convenient relevant examples of how often CGG would be used in modern RNA synthesis, specifically of stretches coding for portions of the SC2 spike protein used in the Pfizer and Moderna vaccines. “The designers of both vaccines considered CGG as the optimal codon in the CGN codon family and recoded almost all CGN codons to CGG.” 19 of 41 Arg codons in Pfizer are CGG, as are 39 of 42 in Moderna. Clearly neither designer used independent choices for different sites but rather each chose one or two favorite codons for repeated use. They were not inspired to use CGG by its appearance in the FCS on the target protein, since none of the other 40 Arg’s on that protein use CGG. Deigin has pointed out another reason that a researcher inserting coding for ArgArg might specifically choose CGGCGG— it provides a marker for a standard, easy, restriction enzyme test allowing the researcher to know if that insertion is still present or has been lost.
The amino acid sequence of the SC2 FCS is identical to a familiar human amino acid sequence that would be a good candidate for use in a furin cleavage site promoting infectivity. In that human FCS sequence the ArgArg pair is coded CGUCGA, which would become CGGCGG either under the choices used in the vaccine codes or to implement the standard tracing procedure described by Deigin.
On the other hand, in the one example of which I’m aware in which a collaborator of the WIV group added a 12nt FCS to a virus (reminiscent of the 12nt addition in SC2) they only used CGG for one of its three Arg’s. This sample is too small to get good statistics on the frequency of CGG coding but it does indicate that the codons are at least partially independent under LL. Based on this one instance, we could give CGGCGG a probability as low as (1/3)2 under LL.
I’ll use the geometric mean between the minimal estimate (0.04) and the high estimate from Moderna (0.93) giving
P(CGGCGG|LL) = 0.19.
The mid-range estimate for the likelihood ratio for updating the odds P(CGGCGG|LL)/P(CGGCGG|ZW) is then 0.19/0.0007 = ~270. Reasonable arguments might raise or lower this by a little more than a factor of 3, so ln(3)=1.1 is reasonable for the standard error of the logit.
Logit3 = 5.6 ± 1.1.
Pre-adaptation
Several other simple features of SC2 would be expected under DEFUSE-style LL but have been widely noted as surprising under ZW. One feature is that the ACE2 binding site worked better for humans than for bats, even before having a chance to evolve in people. As a Nature paper noted “Conspicuously, we found that the binding of the SARS-CoV-2 S protein was higher for human ACE2 than any other species we tested, with the ACE2 binding energy order, from highest to lowest being: human > dog > monkey > hamster > ferret > cat > tiger > bat > civet > horse > cow > snake > mouse.“ The binding to human ACE2 is also substantially stronger than to raccoon dog ACE2. It would also be expected after serial respiratory passage through lab mice with humanized ACE2.
Another relevant feature is that a key binding site of the original SC2 evolved little when grown in human cell cultures. The sharp contrast with the behavior of SARS-CoV-1 strongly suggests that SC2 had already gone through such cell culturing. Likewise, the initial protein evolution in humans was much slower than for SARS-CoV-1, with about a factor of 5 lower ratio of non-synonymous to synonymous mutations. One of the most prominent advocates of the ZW account, Proximal Origins coauthor Eddie Holmes, in a communication with the others on 2/10/2020 noted this contrast with SARS-CoV-1, whose natural origin is established: “It is indeed striking that this virus is so closely related to SARS yet is behaving so differently. Seems to have been pre-adapted for human spread since the get go.”
One might speculate that the slow early evolution in humans was due to some special generalized cross-species infectivity of SC2. That possibility was checked in detail by comparison with early evolution in minks after spillover from humans. The finding was again a sharp contrast between the apparent pre-adaptation for humans and the rapid evolution after spillovers to minks: “[SC2’s] apparent neutral evolution during the early pandemic….contrasts with the preceding SARS-CoV epidemics….Strong positive selection in the mink SARS-CoV-2 implies that the virus may not be preadapted to a wide range of hosts.”
These combined initial adaptation features, each expected for a DEFUSE-style LL but surprising for a ZW origin like that of SARS-CoV-1, should shift the odds further toward LL. Unlike the previous updates they do not easily lend themselves to semi-quantitative form but I think it is hard to see why such features would strike even expert advocates of ZW as anomalous if they were nearly as consistent with ZW as they obviously are with LL. I think that another likelihood factor
P(adaptive features|LL)/P(adaptive features|ZW) = ~3 would be highly conservative. I will use a small standard error only to indicate that much smaller values are implausible, not to imply that much larger values are implausible.
Logit4 = ~1.1 ±0.5
Pre-adaptation combined with intermediate hosts
In treating P(adaptive features|ZW) and P(no wildlife host found|ZW) as independent factors I have made an approximation that overestimates the likelihood of ZW. A virus that circulates extensively in some post-bat wildlife has a chance to evolve from bat intestinal propagation to the very different respiratory propagation mode found in humans, civets, etc. That possibility, however, is nearly ruled out by the failure to find any proximal wildlife host. Even more surprising, no experiment has shown that the initial HSM strain of SC2 is even able to sustainably propagate in raccoon dogs or any other candidate host.
Spillover from sparse wildlife hosts is possible, but that would imply little chance for evolution since leaving bats. The combined data are then less compatible with ZW than would be calculated from a simple product of separate adaptation and host factors. This tension between the limited chances for post-bat pre-human evolution and the apparent pre-adaptation was a topic of discussion among Proximal Origins authors on 2/3/2020. Holmes wrote “No way the selection could occur in the market. Too low a density of mammals: really just small groups of 3-4 in cases.” Garry replied “That is what I thought as well…”. Calculating such potentially major upward refinements of the odds estimate would require some detailed modeling. It is not really necessary for our conclusion here.
Summing up
The key points of the discussion so far are summarized in Table 1.

Summing up our logits and calculating the square root of the sum of their squared uncertainties gives us
Logit = 8.1 ±sqrt (7.6) = 8.1 ±2.8
The point estimate of the logit would correspond to extreme odds, P(LL)/P(ZW) = ~3300. Consideration of the uncertainty in the estimate of the logit will bring those odds down substantially. The reason is not hard to see. If our point estimate of the logit, corresponding to P(LL) = ~99.97%, is low, raising it picks up almost no extra P(LL) because it’s already almost 100%. If on the other hand we were to lower our logit point estimate there is plenty of room for P(LL) to go down.
Let’s crudely estimate how the uncertainty in our estimated factors reduces the net odds by approximating the probability distribution for the sum of the logits by a Gaussian. Numerical integration over the resulting distribution (See Appendix 3) gives
P(ZW)=0.0076
That corresponds to odds of ~130/1. If instead of a Gaussian we used a common fat-tailed distribution, a 3-degree-of-freedom t-distribution, that would decrease the odds to ~100/1. These odds estimates are in-between the previous attempts at comprehensive quantitative Bayesian estimates, described in Appendix 1, which gave ~30/1, ~500/1, and 1000/1.
I think ~100/1 is conservative because I was conservative about each factor, left out some potentially important other factors that tend to support LL, and allowed reasonable standard errors for the factors. Nevertheless, people tend to underestimate uncertainties, so a reader might well suspect the standard error of the logit should be larger. Increasing the standard error of the logit can pull the odds back toward 50-50 although it cannot reverse that the odds favor LL.
What if I have substantially underestimated the uncertainty, by a factor of two in the variance of the logit? The corrected odds (using a Gaussian) would still favor LL by a factor of 33. What if I’ve massively underestimated the uncertainty, by a factor of two in the logit’s standard error? The corrected odds would still favor LL by a factor of 12. What if despite trying to err the other way I’ve unintentionally overestimated the LL-favoring factors by a huge factor of e4= 55? The corrected odds would still favor LL, by a factor of 8.5. What if I have made both those huge errors? The corrected odds would still favor LL by a factor of over 3. The bottom line is just that LL looks at lot more probable than ZW, with room for argument about exactly how much more probable.
Retrospective on methods
How then could so many serious scientists have concluded that P(ZW) is bigger than P(LL) or even that P(ZW) is much bigger than P(LL)? There was of course a great deal of intensely motivated reasoning, as the recently published internal communications among key players vividly illustrate. For those just following the literature in the usual way, the impression left by the titles and abstracts of major publications suggested that ZW had been confirmed, although we’ve seen that the arguments in the key publications disintegrate or even reverse under scrutiny.
There has also been a familiar methodology problem among the larger community that accepted the conventional conclusion. Although simple Bayesian reasoning is often taught in beginning statistics classes, many scientists have never used it and fall back on dichotomous verbal reasoning. The initially more probable story, ZW in this case, is given qualitatively favored status as the “null hypothesis”. Each individual piece of evidence is then tested to see if it provides very strong evidence against the null. If the evidence fails to meet some high threshold, then the null is not rejected. It is a common error to then think that the null has been confirmed, rather than that its probability has been reduced by the new evidence. After a few rounds of this categorical reasoning, one can think that the null has been repeatedly confirmed rather than that an overwhelming likelihood ratio favoring the opposite conclusion has been found.
What should be done?
Despite prior probabilities favoring zoonosis we have seen that after evidence-based updating the odds strongly favor a lab leak origin. How might that inform our actions?
Blaming China is about the most counterproductive possible reaction. The lead Proximal Origin author, Andersen, alluded to the dangers of such blame when on 2/1/2020 he asked his colleagues: “Destroy the world based on sequence data. Yay or nay?” We’ve now seen what the sequence data say but we don’t want to destroy the world— just the opposite. We need to regulate pathogen research in ways that avoid the most dangerous work while expanding work needed to develop vaccines and therapies. No new ideas are needed for the guidelines, since in 2018 Lipsitch already outlined exactly the sort needed to achieve those goals. Meanwhile, paying attention to lab risks cannot be an excuse to ignore ongoing zoonotic risks.
Reflection
None of the three clear existential threats to humanity– global warming, new pathogens, and nuclear war– can be addressed without science. I think that some public trust in science is a necessary though not sufficient condition for successful defenses against those threats. For example, public awareness of the scientific conclusion that SC2 mainly spreads by aerosols and of the value of indoor air filtering would have limited and still could limit the disease burden. When scientists are not candid about what we know we undermine the necessary public trust.
Appendix 1: Previous Bayesian analyses
Demaneuf and De Maistre’s Bayesian analysis, written before DEFUSE was known and omitting sequence considerations, provides a useful introduction to the form of the arguments, as well as detailed analyses of the priors. Readers who find something confusing about the basic reasoning may find their “rebuttal of common misunderstandings” particularly useful.
A brief Bayesian analysis by J. Seymour only considering priors and geographical factors (like my early one) came out in Jan. 2021. It considers a range of possible values obtaining estimates of lab leak probability ranging from 0.05% to 91%. The biggest difference from my current analysis is that Seymour uses no biological data, but he also mostly uses lower priors, without empirical explanation.
The first fairly comprehensive Bayesian analysis that took geographical, biological, and social factors into account came out in 2020 from “rootclaim”. It concluded that some lab event is about thirty times as likely as a pure zoonotic wildlife scenario. That analysis contains a wealth of useful references and discussion but is a bit out of date and uses an obscure method of accounting for uncertainties in the factors.
An extraordinarily detailed analysis from early 2021 by S. Quay concluded that the probability of a lab leak origin was 99.8%, i.e. 500 times as likely as pure zoonosis. (I had forgotten hearing of Quay’s paper until after I finished the core analysis of this paper, so the detailed analyses are independent.) Although there is overlap with my analysis, Quay’s mathematical treatment does not follow a systematic logical system, as Andrew Gelman noted.
Louis Nemzer tweeted an analysis on 10/28.2021 that used straight Bayesian methods rather than robust Bayes, i.e. did not include uncertainties on the factors. This analysis is particularly compact and easy to follow. It includes priors that are somewhat less favorable to LL than mine, a large factor that I don’t use for the existence of the FCS, and a smaller factor for the CGGCGG. He does not include factors for non-observation of hosts or for pre-adaptation. Nemzer ends up with 1000/1 odds favoring LL. Since his method is straight Bayes, those odds would correspond to the 3300/1 odds I get before averaging over the plausible distributions of factors.
An anonymous twitter user posted a brief Bayesian evaluation on 6/20/2022 with fairly much overlap with mine, also concluding that a lab leak was much more probable than competing hypotheses. They used the presence of the FCS in a way that I think is not justified, but they do not get around to using some other details of the genomic sequence that I find to be important.
Another anonymous twitter user has posted a handy Bayes calculator that readers can use to make their own estimates. It is suited only for straight Bayes calculations. In order to realistically allow for uncertainty in the factors (i.e. to use robust Bayes) users will need to try various combinations of plausible values and then take a weighted average of the resulting probabilities, not of the resulting odds, to get their best odds estimate.
Appendix 2: Pekar et al.
Although the Pekar et al. analysis has been superseded by analyses based on more complete data, it’s worth looking at it in detail just to get a feel for the reliability of major work in this field. We’ve seen that calculating Bayesian odds involves both picking priors and calculating likelihood ratios. Oddly, after much complicated model-dependent analysis of the likelihood ratio for two spillovers vs. one spillover the prior odds were just arbitrarily assigned to be 1.0. (See page 13 of the Supplement to Pekar et al.) In effect the prior probabilities used for N, the number of successful spillovers, were P(1) =1/2, P(2)=1/2, P(3)=0, P(4)=0, etc. This looks like a post-hoc attempt to inflate the prior probability of N=2.
Let’s assume, pretty realistically, a Poisson distribution for N with expectation value x. We don’t know x but it can’t be very small because then no spillovers would have been found or very big because then even more than two would have been found. A standard non-informative form for the probability density function of x is 1/x. Its integral diverges weakly but that divergence will not affect the odds. We can then easily integrate the Poisson probabilities over x to get the prior odds, P(N=2)/P(N=1) =1/2. (Extension of this method to higher N gives a very weakly divergent sum of probabilities that stays finite if truncated, e.g. at N= population of Wuhan.) It is peculiar that the paper did not use such a simple conventional exercise to obtain the prior odds without post-hoc adjustment.
Three pubpeer analyses find multiple errors in the code used. One seems to be due to a simple copy-paste mistake. The next is somewhat more conceptual, an incorrect normalization of the likelihoods. Together those two “combined corrections reduce the Bayes factors from ~60 to less than 5.” The third is a double-counting error: “Removing the duplicated likelihoods reduces the Bayes factors by a further ~12%.” When also combined with my conventional non-informative priors, the resulting posterior Bayes odds would be just under 2.2.
This remaining small effect depends critically on the quality of the epidemiological model. Fundamental problems with the model have been noted. The simplifications used in the model have been described as strongly inappropriate for SC2, omitting the short-time superspreading events that are typical for SC2. The model used was originally developed for HIV, which has a much different time course. Brief superspreading events make the observed phylogenetic pattern more consistent with a one-spillover picture than it would seem to be in the model used. Allowing for missing data has a similar effect.
The Bayesian modeling exercise in Pekar et al. thus leaves P(N=2) not far from P(N=1). Since whether N=1 favors LL or ZW is unknown this conclusion would not lead us to change our P(LL)/P(ZW) odds even if the phylogeny account had not been superseded by ones based on much more complete data.
Appendix 3: Calculations
The calculations here are not intended to imply unrealistic precision. They are meant simply to use defined logical algorithms to avoid unnecessarily adding even more subjective steps.
To estimate the expected logit and its variance for an event based on observing it M times out of N trials, I subjectively assume a uniform prior on the probability, x, for not finding a host when there actually is one, giving analytically solvable integrals:
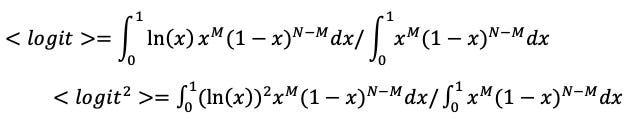
For N=2, M=0 we get <logit> = -1.67 with standard error of 1.59.
For N=4, M=1 we get <logit> = -1.28 with standard error of 0.68.
For N=9, M=2 we get <logit> = -1.43 with standard error of 0.55.
To see what we would get if we dropped the independence assumption for the two CGG’s under ZW and instead used that there were zero CGGCGG’s out of > 3000 ArgArg’s, this method (uniform priors on the probability) gives almost exactly (this and remaining integrals are done numerically)

This would disfavor ZW substantially more than the ln(1/1400 ) = -7.24 that we used assuming independence. Since observation of zero is reasonably likely under the independence assumption (p = ~ 0.12) I conservatively stick with the independence assumption.
To calculate P(ZW) I integrate the probability 1/(1+ex) of ZW for a value of logit = x, over a probability distribution for x with mean L and variance V obtained from the estimates given for the individual logit contributions.

Appendix 4: FCS uses
The FCS appears at several points in the argument, so it may help to clarify in what ways it is used and in what ways it isn’t used.
Although some have argued that having an FCS is very unlikely for this type of coronavirus, that low likelihood may not apply when one remembers the precondition that we wouldn’t be discussing this virus if there weren’t a pandemic for which the FCS may be nearly needed. So I allow P(FCS|ZW, pandemic) to be close enough to 1 to ignore.
Wuhan is not the only place where pathogen research is done, so a priori it would be an exaggeration to say P(Wuhan|LL, pandemic) = ~1. However, the combination of the DEFUSE proposal to add an FCS to coronaviruses, along with other DEFUSE proposed features found, strongly indicate that if SC2 originated from a lab, it would be one doing the DEFUSE-proposed work. The site mentioned in DEFUSE for adding an FCS to a coronavirus, UNC, is smaller and uses highly enhanced BSL3 protocols. After DEFUSE was not funded, switching this part of the work to WIV, where there was already expertise in the methods, would have been easy. A note from a lead investigator, Peter Daszak, to the NIH about earlier work had assured them in 2016 that “UNC has no oversight over the chimera work, all of which will be conducted at the Wuhan Institute of Virology.” While the chance of a spillover occurring at UNC isn’t zero, it’s much lower than for WIV. Thus P(Wuhan|LL, coronavirus with FCS, etc.) = ~1.
The detailed contents of the FCS, the CGGCGG sequence, provide one of the key pieces of evidence used, since P(CGGCGG|LL) >> P(CGGCGG|ZW).
Deigin points out that FCS in SC2 occurs exactly at the S1/S2 junction, as proposed in DEFUSE. Since that is an evolutionary advantageous location, it might only provide a small update factor favoring LL, which I don’t use.
The neighborhood of the FCS, differing from related viruses only by synonymous mutations, has been cited as evidence for LL because it looks peculiar under ZW but not under LL. One of the Proximal Origins authors, Robert Garry, initially reacted: " I really can't think of a plausible natural scenario where you get from the bat virus or one very similar to it to [SC2] where you insert exactly 4 amino acids 12 nucleotide that all have to be added at the exact same time to gain this function -- that and you don't change any other amino acid in S2? I just can't figure out how this gets accomplished in nature. Do the alignment of the spikes at the amino acid level -- it's stunning. Of course in the lab it would be easy to generate the perfect 12 base insert that you wanted.” I don’t use this evidence. Enough is enough.
fantastic work!
I agree with almost everything, just a few comments:
- I'd put some weight on the earliest genome in presence of Vero DNA sequenced at Sangon. Both the company and Vero cell lines are mentioned in DEFUSE. If it were from a human, it would have likely been a less mixed sample, and was certainly never published, which in itself is suspicious.
IMO WIV panick-sequenced all their RaTG13-like / FCS insertion project samples after they got the first SARS2 sequence to check if it was them. This would also explain the CHO DNA (often used for spike characterisation).
- There are 2 more important pieces of evidence you do not discuss
1: human optimized SARS2 spike expression vectors found in 2019 patient samples
2: our endonuclease preprint, specifically the high concentration of syn. Mutations in restriction sites used by WIV researchers in 2017.
this talk may help:
https://youtu.be/EuuY94tsbls?si=IVu6DXPxMDxhNT98
let me know if you like to discuss this.
I don't want to censor anybody, especially one of my rare cohort of fellow Harvard people who did time in a penitentiary. Nonetheless, I'm deleting a series of very long Comments from reader "Harvard2TheBigHouse" because they wandered off topic into extremely naive remarks about quantum mechanics, etc. (Next week I'll post something about quantum mechanics!) I don't want this substack to be a woo forum.
His key relevant point was that he believes that SC2 came from live-attenuated-vaccine research, which I would consider to be a subset of LL. He gave a link to an early paper on that: https://onlinelibrary.wiley.com/doi/10.1002/bies.202100017.
He also believes that HIV came from similar research, a topic about which I know nothing.
Readers who wish to follow up on his thoughts may go to the substack under that name.